
Polishing a PacBio Mosquito Genome
I just completed a basic comparison between the two algorithms, Quiver and Arrow, that are used for polishing a PacBio denovo genome assembly. After discussions with a collaborator, I thought it might be valuable to post this information, in case anyone is looking for details on the two algorithms in the future. One interesting finding in my comparisons was the degree to which polishing the genome can improve completeness as measured by ortholog presence, which I detail below.
As a component of my research on Culiseta melanura, I am generating a draft genome of this mosquito, relying on the long reads from the Pacific Biosciences RSII for denovo genome assembly. Long read technologies can be useful in genome sequencing projects where the genome of interest contains repetetive elements, as is the case in mosquitoes. However, a primary consideration in using long read sequencing methods like PacBio is the high native error rate of the reads compared to other sequencing methods. Long reads can be error corrected to very low error rates, either using reads from a higher accuracy sequencer (e.g. Illumina and a hybrid assembly method), or with high enough raw read coverage during assembly, and further error correction of the assembly during subsequent polishing.
In the case of a PacBio-only assembly, two polishing algorithms are available: Quiver and Arrow. Quiver is being phased out in favor of Arrow, which is apparently easier to develop, but a white paper on Arrow is not yet available. Use of these polishing methods for error correction can lead to a final assembly with Q-scores between Q30 and Q60, depending on raw read coverage. Both algorithms are available via Pacific Bioscience’s GenomicConsensus module, which can be installed either as part of the larger SMRT-Link suite, or through PacBio’s Pitchfork utility for just the components needed to polish. I chose the latter.
The Culiseta melanura draft genome used for this project was assembled with Canu v1.6 from 40 SMRTCells and ~30X raw coverage, yielding an assembly of 1.278 gigabases over 25,500 contigs, with a contig N50 of 93.23 kilobases. Polishing with Quiver and Arrow provided modest gains to N50 and slight increases to total genome size. The Quiver-polished assembly was 1.279 gigabases, with a contig N50 of 93.29 kilobases. The Arrow-polished assembly was a total of 1.281 gigabases, and a contig N50 of 93.36 kilobases.
While polishing provided only minor improvements to contiguity, the real benefit to polishing PacBio denovo assemblies is in the error correction of reads. One way to evaluate this is with a Benchmarking Single Copy Ortholog (BUSCO) analysis. A BUSCO analysis is a good way to assess genome completeness, leveraging a well-curated database of single copy orthologs to evaluate the completeness of a genome by comparing genes from the genome in question to a single copy ortholog catalog. These catalogs are developed from multiple genomes spread across a given taxonomic group, and are expected to be present in any genome within that group. For instance, all Diptera should have all or nearly all of the 2799 orthologs in the Diptera catalog. Thus, the more complete a genome, the larger the proportion of SCOs we would expect to find using BUSCO assessments. However, if a genome had random sequencing errors that occur in gene regions - as we might expect in the case of an unpolished draft genome - BUSCO might fail to detect orthologs that were represented in the genome, or classify those orthologs as fragmented. Thus, we might expect polishing to increase the BUSCO scores of a genome.
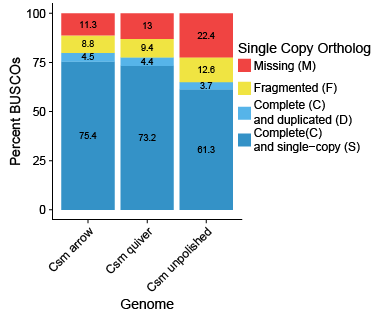 This is in fact what I found. The BUSCO profile for the unpolished, draft genome of Culsieta melanura had only 61.3% complete and single copy universal orthologs from the Diptera catalog, and 22.4% of orthologs were missing (figure at left). Polishing with both Quiver and Arrow increased the relative completeness of the genome, indicating that either some orthologs in the unpolished genome had read errors that caused an ortholog to fail to be detected, or that the additional sequence data in the polished genomes increased BUSCO scores. I suspect it is primarily the former, as the relative change in genome size was very small (less than 1% in both cases), but there was a noticable increase in recovered SCOs between the unpolished genome and polished genomes and between Quiver and Arrow. The Arrow algorithm appeared to perform the best, finding at least partial evidence of 88.7% of SCOs. Interestingly, iteratively running either Quiver or Arrow did not improve genome contiguity or BUSCO scores (data not shown).
This is in fact what I found. The BUSCO profile for the unpolished, draft genome of Culsieta melanura had only 61.3% complete and single copy universal orthologs from the Diptera catalog, and 22.4% of orthologs were missing (figure at left). Polishing with both Quiver and Arrow increased the relative completeness of the genome, indicating that either some orthologs in the unpolished genome had read errors that caused an ortholog to fail to be detected, or that the additional sequence data in the polished genomes increased BUSCO scores. I suspect it is primarily the former, as the relative change in genome size was very small (less than 1% in both cases), but there was a noticable increase in recovered SCOs between the unpolished genome and polished genomes and between Quiver and Arrow. The Arrow algorithm appeared to perform the best, finding at least partial evidence of 88.7% of SCOs. Interestingly, iteratively running either Quiver or Arrow did not improve genome contiguity or BUSCO scores (data not shown).
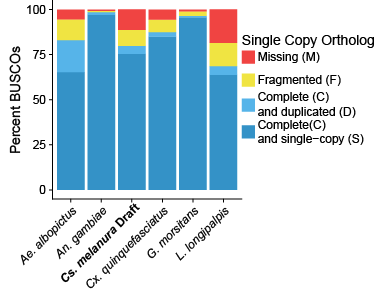 Compared to a few other publicly released genomes on VectorBase, the draft genome of Culiseta melanura is a little less complete (figure at right). However, as a starting place for future sequencing efforts, the genome is in a good place. We are still hoping to complete additional sequencing in the coming months before I finish at the Connecticut Agricultural Experiment Station, and so I will probably provide an updated blog when that happens.
Compared to a few other publicly released genomes on VectorBase, the draft genome of Culiseta melanura is a little less complete (figure at right). However, as a starting place for future sequencing efforts, the genome is in a good place. We are still hoping to complete additional sequencing in the coming months before I finish at the Connecticut Agricultural Experiment Station, and so I will probably provide an updated blog when that happens.
Although Arrow is not yet described as thoroughly as Quiver, it does appear from my results that, at least in the case of the highly repetitive Culiseta melanura genome, it provides slightly better results than Quiver. The Arrow polished genome had a better BUSCO assessment and minor increases in genome size and N50 relative to the Quiver polished genome. Once additional sequencing is completed, I intend to run the above comparison again, to see if additional data tilts in the direction of one algorithm or the other.