
Mini-Assemblathon for Anchor Hybrid Enrichment
As sequence capture methods increase in popularity for phylogenomics, identifying the best tools for use with such methods is becoming ever more pressing. A key step in most sequence capture pipelines is the assembly of loci, and generally, most groups rely on existing software used for denovo genome or transcriptome assembly. I’ve decided to evaluate software options for use with anchor hybrid enrichment, and summarize those results below. I’ll start with a brief overview of the capture methods used in the Wiegmann lab, and follow that with my “assemblathon”.
A quick overview of Anchor Hybrid Enrichment
As part of an NSF-funded project to better understand the evolutionary relationships of Culicidae, I am using a ‘reduced representation’ technique called anchor hybrid enrichment. So-called reduced representation methods aim to reduce the amount of sequencing done per individual by selecting homologous regions of the genome for sequencing. This can make sequencing projects affordable with hundreds of individual samples, particularly in non-model organisms where low coverage genome sequencing would still be intractable due to lack of a full genome reference. RADseq does this with restriction enzymes, while anchor hybrid enrichment, ultra conserved elements (UCEs), and related methods do this with probes that target conserved areas of the genome. 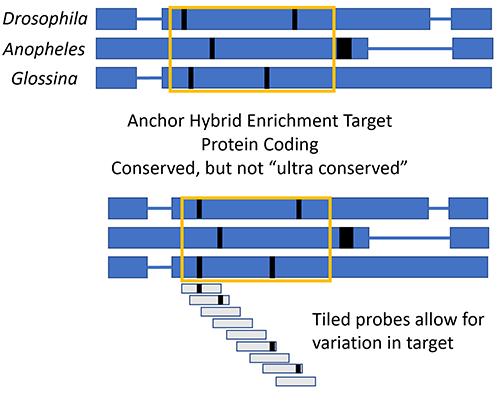
In the case of anchor hybrid enrichment, 120 bp probes are tiled along the conserved - but not ultraconserved - target protein coding sequences (schematic at right). By leveraging tiled probes, genes can be captured that vary in some regions from the references sequences initially targeted, allowing for wide taxonomic coverage, but also enabling a focus to be on the regions being targeted, rather than only the flanking regions as in other sequence capture methods. During wetlab work, we se probes to capture our target regions (as well as similar bycatch) and construct a typical NGS sequencing library from them. We are able to multiplex multiple individuals together, and use any of the various Illumina sequencing platforms to generate our sequence data from these libraries.
Assembling AHE Contigs and the Assemblathon
Next comes the fun part - taking the raw reads and assembling them into something useful - contigs we can use for orthology assessment! To do this, our lab has traditionally used Trinity, due to its excellence in working with gene fragments. Although AHE data are not the same as transcriptomes, they are similar enough that the community has generally stuck with Trinity for assembling sequence capture data (including UCEs).
Recently, however, I explored if other assemblers could be an option - particularly SPAdes, which is a bit of an all-star when it comes to assembling smaller genomes. And so, I set out on a mini assemblathon, testing three different strategies for assembling AHE data with three different samples. First is the tried and true method of Trinity with default settings. Second is SPAdes with default settings. Finally is an assembly strategy using Abyss with four Kmer sizes between 26 and 96, followed by BBmap to merge redundant and overlapping contigs (hereafter AbyssKmers).
The Assemblathon
I selected three samples of varying for use in this preliminary test of assemblers. The three samples had varying input quantities - this is reflected in differences in both starting DNA and in reads, although that is not always representative of the total mosquito genes found in samples (See T1/T2 below). Next, I generated typical statistics used for evaluating assemblies, such as the total number of contigs (Total N), the number above 500 basepairs, the N50, the maximum contig size, and the total sum of all nucleotides on contigs. I also passed each through an ortholog pipeline that identifies phylogenomic markers, and calculated some basic summary statistics from those results. I also evaluated the level of duplication in the assembly, here defined as contigs that share more than 99% identity with one another. This is not a perfect measure of duplication, and may also indicate heterozygosity; in future tests, I’ll try to adjust for that. But for now, this provides us a bit of a starting point.
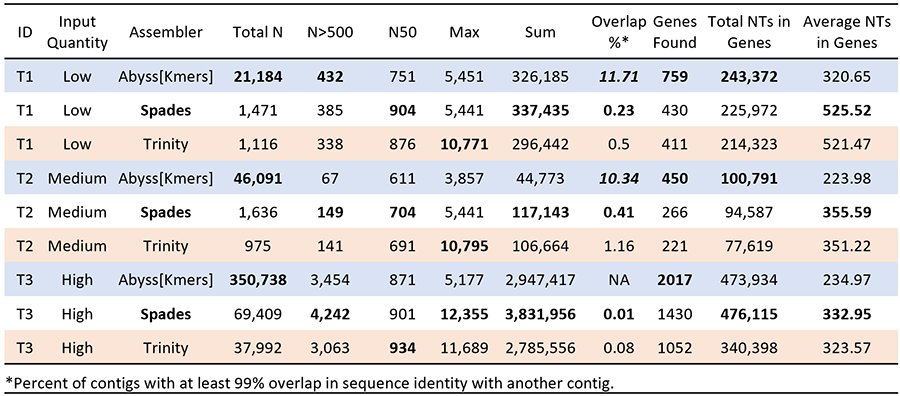
The results of this mini-assemblathon support that SPAdes is likely the best choice for AHE loci assembly - something I believe that UCE folks have also concluded recently. SPAdes does the best when it comes to the total nucleotides across all samples, has the least evidence of duplicate contigs, in general has longer contigs, and in general produces larger genes. AbyssKmers finds by far the most contigs, and the most genes with our orthology pipeline, but they are extremely small on average, which would limit some of their phylogenetic utility. Our filtering steps remove many small loci (particularly those under 100 amino acids, which are quite common in the AbyssKmers assemblies), and so the smaller contigs produced by Abyss are a bit of a concern. As our first 200 AHE assemblies have already been done with Trinity, our move towards SPAdes will likely happen for future publications, but I do think we are going to make that transition in the future.
I’ll be exploring if I can optimize Abyss across multiple Kmers, as well as trying GATB or another pipeline equipped with a similar multi-Kmer approach. And of course, because three assemblies is quite the small sample size, I’ll be repeating this Assemblathon on a bit more data to confirm these results. Regardless, the early results here suggest that SPAdes is likely the best option for these sort of data - not surprising given how great it has been proven to be on smaller genomes.